
Data structures
SeqAn has numerous data structures that are helpful for analyzing biological sequences. Those range from simple containers for strings that can be saved in different ways, to collection of strings or compressed strings.
The Journaled string tree, for example allows the user to traverse all sequence contexts, given a window of a certain size, that are present in a set of sequences.
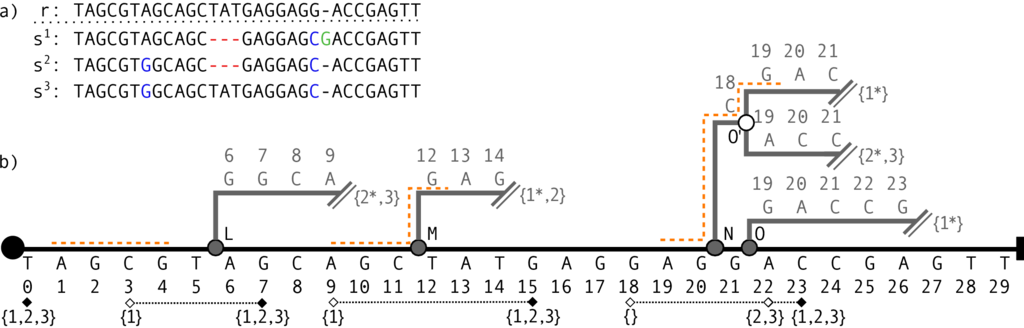
Similar sequences are hence only traversed once, and the coordinate bookkeeping is all within the data structure. This allows for example speedup of up to a 100x given sequences from the 1000 Genome project and compared to traversing the sequences one after another.
Another strong side of SeqAn are its generic string indices. You can think “suffix tree” but the implementations range from an enhanced suffix array to (bidirectional) FM-indices.